

Unpredictability of SCD
Let's change the way we view SCD
LEARN HOW THE ACUTE SYMPTOMS OF SICKLE CELL DISEASE (SCD) COULD LEAD TO CHRONIC COMPLICATIONS

SCD IS A LIFELONG GENETIC DISEASE CAUSED BY A SINGLE MUTATION IN THE HBB GENE THAT LEADS TO THE PRODUCTION OF HEMOGLOBIN SICKLE, HbS
HbS Molecule

Red blood cells (RBCs) of patients with SCD can produce a high concentration of HbS which, in low oxygen conditions, can undergo deoxygenation. When HbS undergoes deoxygenation, it can polymerize and induce distortion, rigidity (causing vaso-occlusion), and structural damage that cause RBCs to be sickle shaped, which become fragile (causing breakdown and shortened lifespan of RBCs). This results in chronic hemolytic anemia and the vaso-occlusive episodes (VOEs) that characterize SCD.1-3

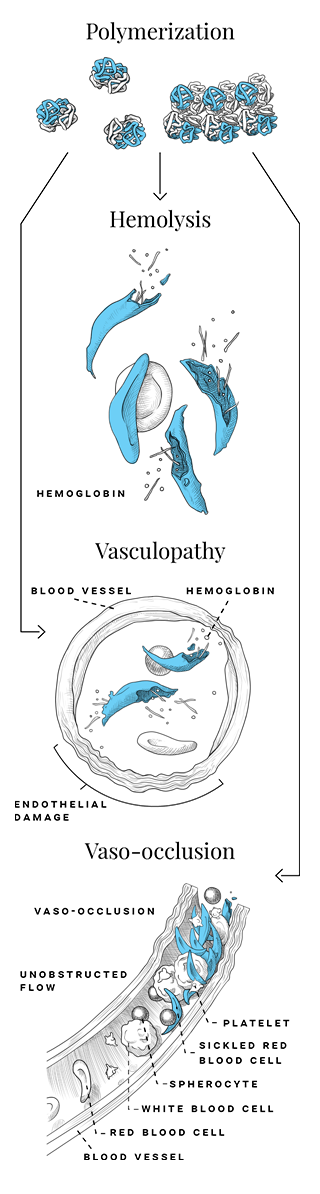
SCD is an inherited lifelong disease that presents with a wide range of disease severity including acute and chronic complications, widespread organ damage, and the risk of early mortality.2
Estimates suggest at least 100,000 Americans are living with SCD4
1 IN 365
newborn Black
Americans will have SCD
1 IN 13
newborn Black
Americans will carry
the sickle cell trait
1 IN 16,300
1 IN 16,300
newborn Hispanic
Americans will have SCD
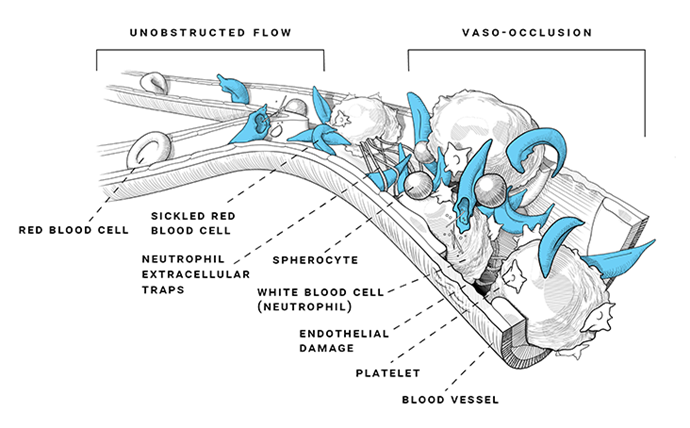
Vaso-occlusive events are the hallmark of SCD
Vaso-occlusive events (VOEs) are a group of acute complications that are associated with SCD and include vaso-occlusive pain crises, acute chest syndrome (ACS), stroke, and splenic sequestration; additional acute complications are also listed in the table below.1,2
VOEs can be triggered by a multitude of factors that cause the deoxygenation of HbS, such as1:

Illness

Dehydration

Cold temperature

Increased wind speed

Higher barometric pressure
However, VOEs can occur without warning and can have a very abrupt and severe impact on patients’ quality of life. Vaso-occlusive pain crisis is the most common VOE and is the leading cause of hospitalization. Pain crises are described differently for different patients—they can present as any type of pain such as aching, drilling, pounding, feel like sharp knives, or throbbing, and can occur anywhere and migrate to other points of the body. Vaso-occlusive pain crises can have a slow or rapid onset, and last a few hours, a week, or even longer. While pain crises are the most common acute complication of SCD, SCD can also cause other unpredictable acute complications. These complications are also important as they can lead to irreversible organ damage and potentially early death.1,5

| Acute complications of SCD | Acute complications of SCD |
| Acute pain from vaso-occlusion (or pain crises) typically felt in the extremities, chest, and back are the primary reason for emergency department (ED) visits. In some cases, vaso-occlusive crises can lead to sudden and unexpected death.1,2 | |
| Acute chest syndrome (ACS) can or will affect approximately 30% of people living with SCD. ACS severity increases with age and more than 10% of adult cases are either fatal or complicated by neurological events and organ failure.2,6 | |
| Infection rates are high in children with SCD and are associated with an increased risk of death from sepsis; infection with parvovirus B19 can result in RBC count drop that may lead to aplasia and require blood transfusion.7,8 | |
| Priapism is a painful, involuntary, nonsexual penile erection that is associated with SCD in 11% to 67% of males living with SCD, although this condition is likely underreported; recurrence may lead to impotence.9,10 | |
| Stroke; overt thrombotic stroke is a VOE that historically most commonly occurred by the age of 10; over time, 1 out of 4 adults living with SCD (25%) will experience a stroke by the age of 45; hemorrhagic strokes account for 3-30% of acute neurological events.2,11 | |
| Cerebral vasculopathy is experienced by about 50% of people living with SCD by the age of 14 and can manifest acutely as overt or silent stroke, as well as cerebral hemorrhage and headaches.11,12 | |
| Splenic sequestration is one of the earliest life-threatening complications (usually at 5 to 24 months of age) in SCD and occurs in 7% to 30% of young children with SCD; untreated, pooling of blood in the spleen leads to a drop in hemoglobin, and can lead to shock and death.2,8,13 |
Look beyond the pain—and see the impact of both acute
and recurrent complications of SCD

The treatment of an individual living with sickle cell disease, I think of it as looking at their whole lifespan. We make sure they understand all the potential complications of SCD.”
Abena O. Appiah-Kubi, MD
Clinical manifestations of SCD may not show the degree of underlying organ damage
Compared with the acute events, less is known about organ damage resulting from repeated vaso-occlusion, infarction, and chronic hemolytic anemia. Organ dysfunction and failure lead to14:
- High healthcare resource utilization
- Poor quality of life
- Shortened survival
Because of the cumulative nature of the disease, chronic organ complications become the main cause of morbidity and mortality by age 30.2,14 Some of the most profound long-term complications of SCD include:
Anatomy
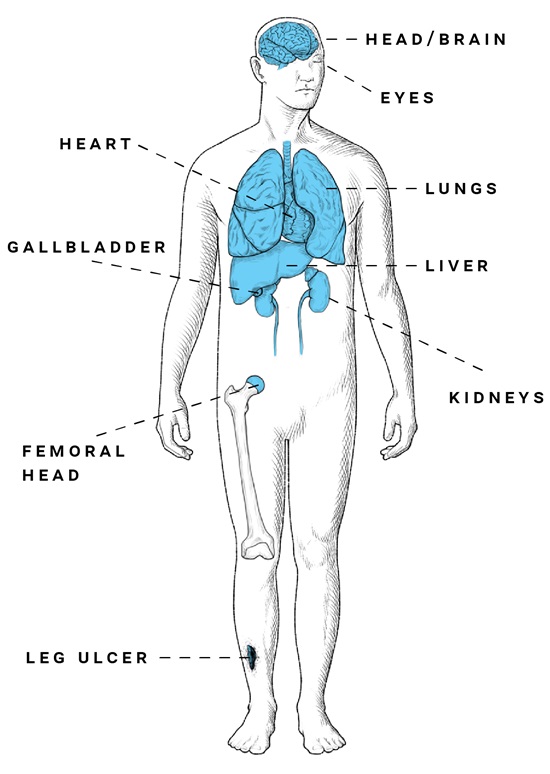
| Long-term and ongoing complications of SCD Long-term and ongoing complications of SCD | |
| Sickle cell retinopathy | 38% and 76% prevalence, depending on genotype; can lead to a loss of visual acuity if left untreated. Because it is often asymptomatic, sickle cell retinopathy requires retinal screening.2,15 |
| Pulmonary hypertension | 6% to 11% prevalence, with an increase in severity related to thromboembolic disease, obstructive sleep apnea, hypoxemia, and an increased risk of death. Pulmonary hypertension results from the effects of hemolysis, which promote clinical complications of vasculopathy.16,17 |
| Cardiovascular complications | Complications increase with age and are a common cause of death for people living with SCD.3 |
| Cerebral vasculopathy | Overt stroke occurs in 11% of patients younger than 19 and in 24% of adults by age 45; prevalence of silent infarcts increases with age from 27.7% before age 5 to 53.5% by age 30, and is associated with neurocognitive dysfunction.18,19 |
| Cholelithiasis | 23% to 66% prevalence in the SCD population.20 |
| Renal dysfunction | One of the most common complications of SCD, which includes renal insufficiency, hyposthenuria, proteinuria, and renal failure requiring dialysis; 1 study reported that the mean time to death in end-stage renal disease is 4 years.21,22 |
| Avascular necrosis of femoral head | Associated with crippling pain in affected people; 22% prevalence, with 23% of these people requiring a hip replacement at a median age of 36, according to a state-wide analysis of 6237 patients with SCD.23 |
| Recurrent, chronic leg ulcer | 18% prevalence in adults with SCD; this is a painful and debilitating complication of SCD and is linked to stroke, ACS, thrombosis, renal disease, and elevated tricuspid jet regurgitation.24,25 |
| Silent stroke (asymptomatic cerebral infarction) | The most common cause of neurologic injury in adults and children with SCD, associated with diminished IQ scores and poor academic performance; an increase in the prevalence of silent infarcts is highly associated with age.19 |
| Depression and anxiety | Affects ~35% of people living with SCD and is associated with worse quality-of-life outcomes; the prevalence is ~5 times greater than that of the general population.26 |
| End organ disease | Occurs in 59% of adults due to chronic complications, with multiple organs or multiple systems often being affected.27 |
Refocus the care of patients with SCD to include active monitoring and long-term disease management
Although acute pain crises are the most common manifestations of SCD, comprehensive care may require more than just treating overt complications. Patient care in SCD may include monitoring progressive damage, managing acute and chronic complications, and planning for long-term care.

It really is day by day of how you're going to feel. So it's hard to really have a normal life like other people. One day I may not feel like getting out of bed, or maybe it takes 2 hours for my legs to stop hurting.”
Artisha, living with SCD

Our partnership is our strength
together for better outcomes in this disease Let’s change the way we
view SCD and work together for
better outcomes in this disease
References
1. Brandow AM, Zappia KJ, Stucky CL. Sickle cell disease: a natural model of acute and chronic pain. Pain. 2017;158(Suppl 1):S79-S84.
2. Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010.
3. Miller AC, Gladwin MT. Pulmonary complications of sickle cell disease. Am J Respir Crit Care Med. 2012;185(11):1154-1165.
4. Centers for Disease Control and Prevention. Sickle Cell Disease (SCD). Accessed April 1, 2020. https://www.cdc.gov/ncbddd/sicklecell/data.html
5. Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120(18):3647-3656. 6. Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. Blood. 1994;84(2)643-649. 7. Adamkiewicz TV, Sarnaik S, Buchanan GR, et al. Invasive pneumococcal infections in children with sickle cell disease in the era of penicillin prophylaxis, antibiotic resistance, and 23-valent pneumococcal polysaccharide vaccination. J Pediatr. 2003;143(4):438-444. 8. Lanzkowsky P, Linton JM, Fish JD. Lanzkowsky's Manual of Pediatric Hematology and Oncology. 6th ed. Academic Press; 2016. 9. Serjeant G, Hambleton I. Priapism in homozygous sickle cell disease: a 40-year study of the natural history. West Indian Med J. 2015;64(3):175-180. 10. Kato GJ. Priapism in sickle-cell disease: a hematologist's perspective. J Sex Med. 2012;9(1):70-78. 11. Brousse V, Kossorotoff M, de Montalembert M. How I manage cerebral vasculopathy in children with sickle cell disease. Br J Haematol. 2015;170(5):615-625. 12. Bernaudin F, Verlhac S, Arnaud C, et al. Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood. 2011;117(4):1130-1140; quiz 1436. 13. Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol. 2014;166(2):165-176. 14. Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390(10091):311-323. 15. van Tuijn CFJ, Schimmel M, van Beers EJ, Nur E, Biemond BJ. Prospective evaluation of chronic organ damage in adult sickle cell patients: a seven-year follow-up study. Am J Hematol. 2017;92(10):E584-E590. 16. Ataga KI, Klings ES. Pulmonary hypertension in sickle cell disease: diagnosis and management. Hematology Am Soc Hematol Educ Program. 2014;2014(1):425-431. 17. Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127(3):750-760. 18. DeBaun MR, Kirkham FJ. Central nervous system complications and sickle cell disease. Blood. 2016;127(7):829-838. 19. Kassim AA, Pruthi S, Day M, et al. Silent cerebral infarcts and cerebral aneurysms are prevalent in adults with sickle cell anemia. Blood. 2016;127(16):2038-2040. 20. van Beers EJ, van Tuijn CFJ, Mac Gillavry MR, et al. Sickle cell disease-related organ damage occurs irrespective of pain rate: implications for clinical practice. Haematologica. 2008;93(5):757-760. 21. Naik RP, Derebail VK. The spectrum of sickle hemoglobin-related nephropathy: from sickle cell disease to sickle trait. Expert Rev Hematol. 2017;10(12):1087-1094. 22. Sharpe CC, Thein SL. How I treat renal complications in sickle cell disease. Blood. 2014;123(24):3720-3726. 23. Adesina O, Brunson A, Keegan THM, Wun T. Osteonecrosis of the femoral head in sickle cell disease: prevalence, comorbidities, and surgical outcomes in California. Blood Adv. 2017;1(16):1287-1295. 24. Minniti CP, Eckman J, Sebastiani P, Steinberg MH, Ballas SK. Leg ulcers in sickle cell disease. Am J Hematol. 2010;85(10):831-833. 25. Minniti CP, Delaney KM, Gorbach AM, et al. Vasculopathy, inflammation, and blood flow in leg ulcers of patients with sickle cell anemia. Am J Hematol. 2014;89(1):1-6. 26. Adam SS, Flahiff CM, Kamble S, Telen MJ, Reed SD, De Castro LM. Depression, quality of life, and medical resource utilization in sickle cell disease. Blood Adv. 2017;1(23):1983-1992. 27. Chaturvedi S, Ghafuri DL, Jordan N, Kassim A, Rodeghier M, DeBaun MR. Clustering of end-organ disease and earlier mortality in adults with sickle cell disease: a retrospective-prospective cohort study. Am J Hematol. 2018;93(9):1153-1160.